

美國FDA 醫材法規最新動態 QMSR :
2. 美國於 2024.01.31 發布 21 CFR part 820(新版) 關於醫療器材品質系統法規 (QMSR) 的最終規則制定(Final rule)。
3. QMSR與國際標準 ISO 13485:2016 進行調和修訂,此修訂法規將於 2026.02.02 正式生效。
4. 進入美國市場的醫材製造商有兩年時間來調和及緩衝,以滿足 QMSR 的要求,於此之前仍須遵守現有的 QSR規範。
FDA : Quality Management System Regulation(QMSR)Final Rule
美國FDA醫材上市前通知510k申請輔導之說明:
製造業者或人士若想將醫療器材(Class I, II, III, De Novo)行銷到美國前,除部分豁免510(k)品項,及無須進行上市前核准(Premarket Approval, PMA)外,必須在進口美國至少90天前向美國FDA提出上市前通知(Premarket Notification, PMN)申請,取得輸入許可函(510(k) Clearance Letter)。上市前通知510(k)是向FDA提交的上市前提交文件,旨在透過證明與合法上市醫療器材(類似品,Predicate Device)具有實質等效性(Substantially Equivalent,SE),證明待上市醫材是安全且有效的。提交者必須將其510(k)醫材與在美國合法銷售的類似設備進行比較。然而,任何合法的美國已上市的設備可以作為類似品。這包括已通過510(k) 流程的器材,1976年5月28日之前合法銷售的器材(修正案前的設備);最初於美國的設備以 III等級器材上市(上市前核准),隨後降級為II二等級或I一等級;或 510(k) 豁免器材。
510k申請註冊輔導之醫療器材等級:
1. 16大類的醫材依照風險等級被歸類成三大等級(CLASS I、II、III)。2. 依照等級不同,醫材的安全性與有效性被FDA管轄的方式也不同。
3. 因此決定醫材等級是申請FDA首要進行的步驟。
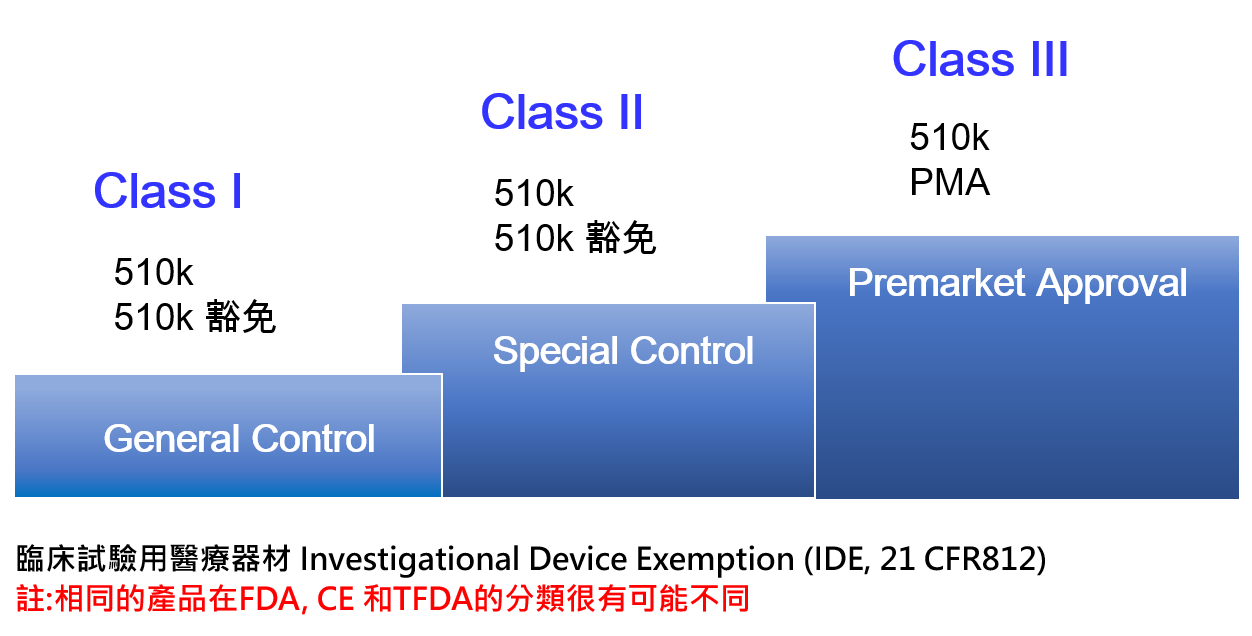
510(k)申請註冊輔導之一般管制 (General Control):
1. 企業註冊
2. 産品列名
3. Premarket Notification (510(k)),unless exempt
4. 品質系統 (QMSR)
5. 醫療器材上市後不良通報 (MDR)
6. 禁止濫造和錯誤的標簽 (Labeling) CFR part 814 部份第三類高風險性醫材無法經由與510(k)核准器材實質等同(SE) 或對於疾病有顯著風險,需進行上市前審查。
510k申請註冊輔導之特殊管制 (Special Control):
除了滿足一般管制外,還有以下額外要求:
1. 上市後監督(Postmarket Surveillance Study)
2. 使用者記錄
3. 強制的性能標準
4. 特殊的標籤規定
5. 指引(Guidance)
美國FDA之上市前審查 (Premarket Approval, PMA):
21 CFR part 814 部份第三類高風險性醫材無法經由與510(k)核准器材實質等同(SE) 或對於疾病有顯著風險,需進行上市前審查。
美國FDA之醫療器材豁免(Exempt):
1. 21 CFR part 812 允許臨床試驗醫材進行臨床試驗,以確認器材的安全與有效性資料,做為未來PMA or 510(k)提交資料。
2. 對於醫材具有顯著風險,廠商須先經由FDA & IRB核准後,才可執行臨床試驗,無顯著風險產品,僅需IRB核准。
美國FDA之新且低風險之醫療器材程序(De Novo):
1. 針對中低風險醫材,但與上市產品不具備實質等同 FDA核准後,將歸類在Class I or II。
2. 若正在申請510(k),收到FDA發出的非實質等同(NSE)後, 業者於30天內向FDA提交De Novo。
3. 或直接提交De Novo,業者確認與上市產品無SE,直接申請De Novo之業者須說明未找到SE或為何不適用PMA。
510k顧問申請輔導要點:
1. 如何搜尋類似品 (predicate device)
FDA 510(k)資料庫包含根據 510(k)流程批准的所有醫材,FDA 網路資料庫每月約 5 日更新。設備和產品代碼的分類對於搜尋類似品至關重要。您可以透過在產品代碼分類資料庫中執行搜尋來找到您的器材的分類。如果您的器材類型已獲得 FDA的最終分類(例如 21 CFR 888.1100:關節鏡),分類資料庫將提供該類別(例如骨科裝置)、通用名稱、產品代碼和CFR法規。
對於尋找510(k)類似品有用的資訊包括:
(1) 類似器材的名稱-器材銷售時使用的商品名稱。
(2) 類似器材的製造商。
(3) 行銷狀態,即法規修訂前或修訂後器材。
(4) 510(k)後修改裝置號碼。
(5) 器材的分類訊息,即產品代碼、分類規定等。
2. 美國FDA註冊(Registration)與登錄(Listing)
註冊與登錄是FDA用來建立醫療器材業者管理資料檔案的機制,根據FD&C Act第510節規定,醫療器材的製造業者(manufacturer)、初始經銷商或進口商(initial distributor or importer)及經銷者(distributor)都必須註冊 (Registration),須向FDA登錄基本資料,假如註冊者有重大變更(更換負責人)時,必須在30天內通知FDA。
再者,醫療器材製造業者必須在產品正式銷售前,向FDA登錄(Listing)其所製造且在美國銷售的全部醫材產品名稱。如果資料有任何更改,同樣必須在FDA網站進行更新。
3. 依據美國FDA規定企業或團體不會獲得任何豁免或減免,所有機構都必須繳納機構註冊費,而若年營業額低於1 億美元以下的小型企業則可依證明文件來減少申請費用,美國FDA要求所有醫療器材製造業者或企業必須在該年度「前一年」的10月1日至12月31日期間完成年度註冊,亦即要完成繳費並在網路完成美國代理人及公司登記,例如2024年度須在2023年10月1日至12月31日期間完成年度註冊。
相關資訊: 美國FDA 公告2026年醫療器材申請費用 / FDA 小型企業申請指引
510k申請註冊輔導之醫材登錄 (MD Listing)
1. 21 CFR part 807 業者須向FDA申請產品列名,對於產品列名,先鑑別產品是否需要510(k)/PMA核准或免除。2. 申請者可為: 醫療器材製造商、合約製造商、合約滅菌者、重新包裝廠商、重新貼標廠商、重新製造廠商、規格開發廠商、單次使用醫材再加工業者、配件&零件製造廠商、外銷醫療器材廠商。
3. 若需要510(k)或PMA,產品登錄只能在獲得SE letter或PMA核准之後才能登錄。
4. 假如該產品是唯一登錄的產品,需在獲得510(k)/PMA再進行廠商註冊。
5. 登錄的醫療器材並不代表企業或器材被FDA批准。
6. 如果510(k)或PMA的批准函上有多個產品編碼,需要在每個產品編碼下進行登錄。
510k申請註冊輔導之何時申請510k
1. 販售新的醫材到美國境內。2. 販售已經合法上市,但有跟先前不同預期使用目的的醫材到美國境內。
3. 已經合法於美國上市,但是後來有經過變更或修改過的醫材。
510k申請註冊輔導之醫療器材分類
依據器材的預期用途和使用方法進行産品分類,共有1700多個類別 (Regulation number),不同類別的器材被歸爲16個大類,稱爲Panel (21 CFR 862-892)862 Clinical Chemistry and Clinical Toxicology(臨床化學與毒理學)
864 Hematology and Pathology(血液與病理科)
866 Immunology and Microbiology(免疫學與微生物學)
868 Anesthesiology(麻醉科)
870 Cardiovascular(心血管科)
872 Dental(牙科)
874 Ear, Nose, and Throat(耳鼻喉科)
876 Gastroenterology and Urology(腸胃與泌尿科)
878 General and Plastic Surgery(一般與整形外科手術)
880 General Hospital and Personal Use(一般醫院與個人使用)
882 Neurology(神經科)
884 Obstetrical and Gynecological(婦產科)
886 Ophthalmic(眼科)
888 Orthopedic(骨科)
890 Physical Medicine(物理醫學科)
892 Radiology(放射線與醫療影像)

 加入領証生醫專頁
加入領証生醫專頁