

QMS 醫材輔導
由於TFDA對於醫療器材產品的安全性與有效性嚴格要求,醫材製造廠在向TFDA提出申請醫材許可證註冊前,必須通過醫療器材品質管理系統準則QMS (Quality Management System)查廠,QMS準則隨母法《醫療器材管理法》2021年05月01日生效,應於2024年4月30日全面符合QMS並取得醫療器材製造許可。為了促進台灣醫材產業快速取得QMS認證,領証資深QMS顧問提供優質QMS申請輔導之專業服務。
ISO 13485 醫材輔導
台灣製造的醫療器材在外銷各國市場前,必須符合當地的醫材法規。由於ISO 13485品質管理系統認證為全球多數國家所採認,領証ISO 13485顧問及醫材法規顧問可輔導台灣醫材製造業者向全球擴展市場,提供ISO 13485顧問輔導諮詢,協助製造業者快速取得ISO 13485醫材品質管理系統認證,致力提升醫材產業行銷海內外之醫材產品及強化競爭力。
ISO 13485 的關鍵要求

QMS申請輔導
QMS稽核作業流程圖
QMS / ISO 13485 顧問輔導要點

QMS及ISO 13485 顧問輔導重點
QMS及ISO 13485稽核常見缺失比率次序為設計管制、採購、檢驗量測及測試設備之管制、檢驗與測試、製程管制等(102年CDE),設計管制、製程管制、品質系統、檢驗與測試、矯正與預防措施、識別與追溯性、管理責任、採購等(94~99年TFDA),可見設計管制是QMS及ISO 13485顧問輔導第一大重點。
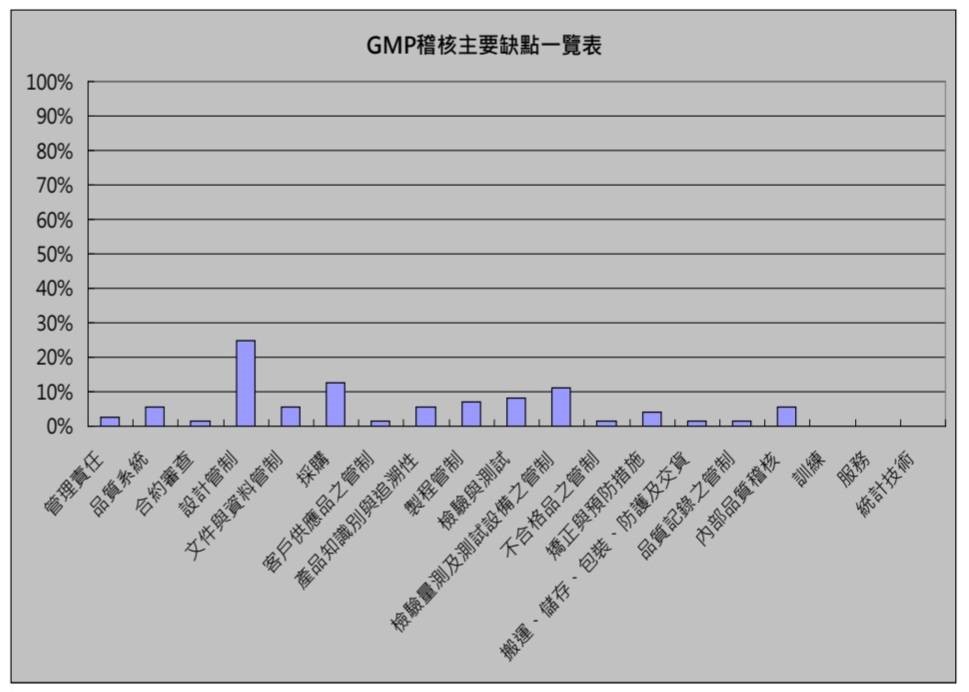
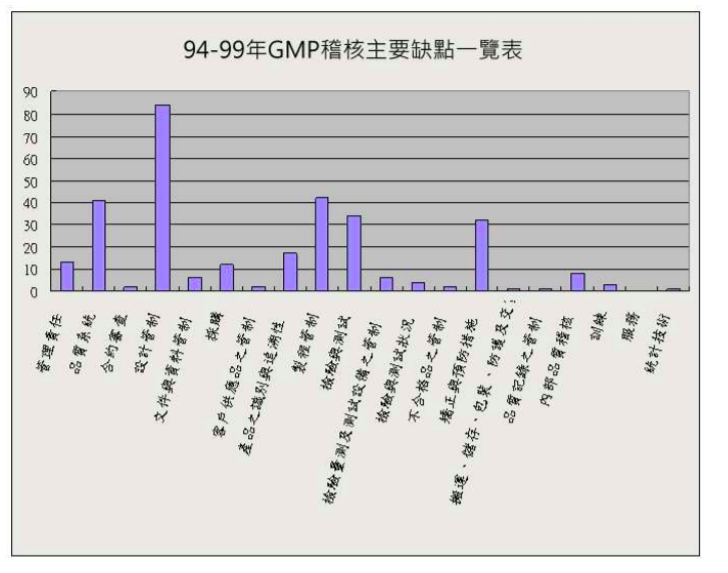
Source: TFDA / CED
QMS及ISO 13485顧問輔導之設計管制
1. 設計管制
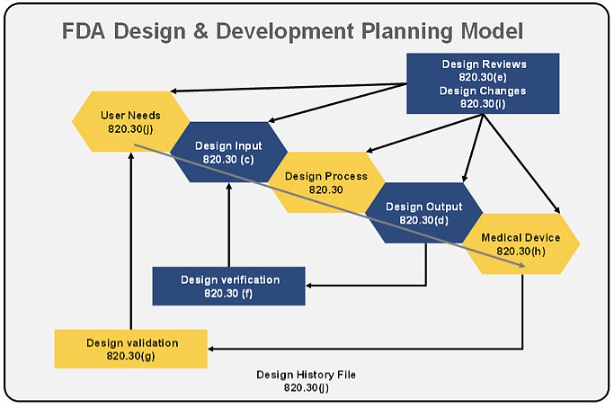
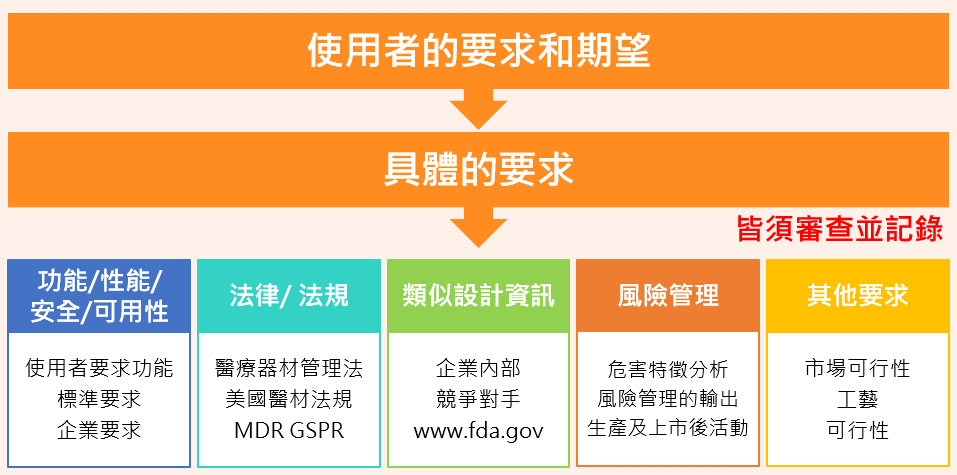
2. 設計與開發輸入
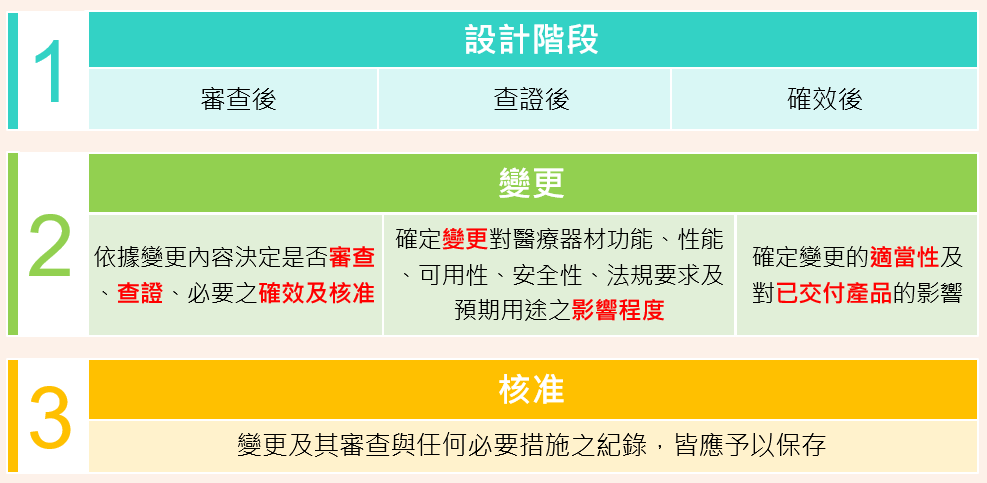
3. 設計與開發查證(驗證)

4. 設計與開發變更
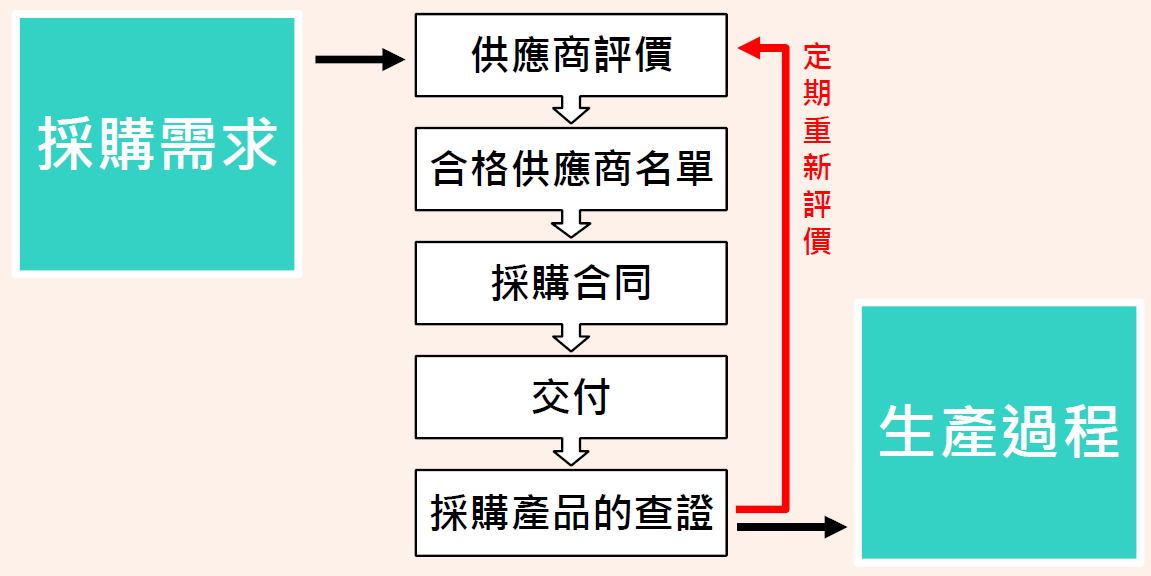
QMS及ISO 13485顧問輔導之採購管制
QMS及ISO 13485顧問輔導之UDI要求
1. 醫療器材管理法於2020年1月15日由總統公布,依據醫療器材管理法第33條訂定”醫療器材標籤應刊載單一識別碼規定” 2023年2月13日生效,第二級及第三級醫材之單一包裝或器材本體上,應標示單一識別碼(UDI);而單一識別碼之產品對應資訊,應至醫療器材單一識別系統資訊管理平台登載(TUDID)。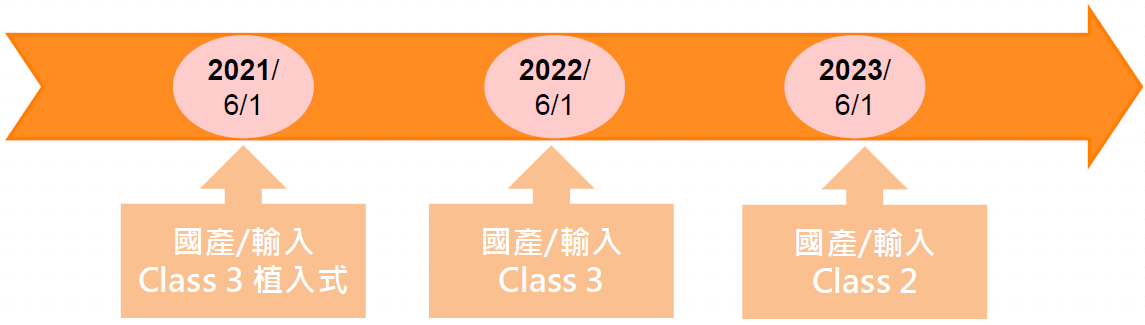
2.歐盟EU UDI之要求:
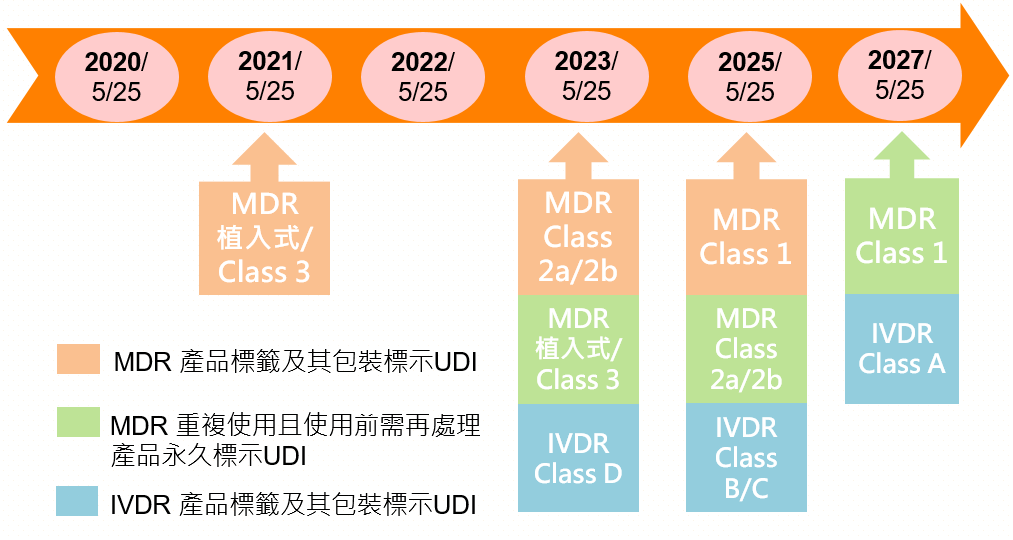
TFDA QMS 申請代辦顧問輔導
國內醫療器材製造業者品質管理系統(QMS)申請:1.QMS法規依據:
(1)醫療器材管理法第二十二條第一項,醫療器材製造業者應建立醫療器材品質管理系統,就場所設施、設備、組織與人事、生產、品質管制、儲存、運銷、客戶申訴及其他事項予以規範,並應符合醫療器材品質管理系統準則(QMS)。醫療器材製造業者依前項準則規定建立醫療器材品質管理系統,並報中央主管機關檢查合格取得製造許可後,始得製造。但經中央主管機關公告之品項,得免取得製造許可。
免取得醫療器材製造許可品項: https://www.fda.gov.tw/TC/newsContent.aspx?cid=3&id=27186
(2)依據醫療器材管理法第10條規定醫療器材管理法所稱「醫療器材製造業者」,指下列二類業者:
(a)從事醫療器材製造、包裝、貼標、滅菌或最終驗放。
(b)從事醫療器材設計,並以其名義於市場流通。
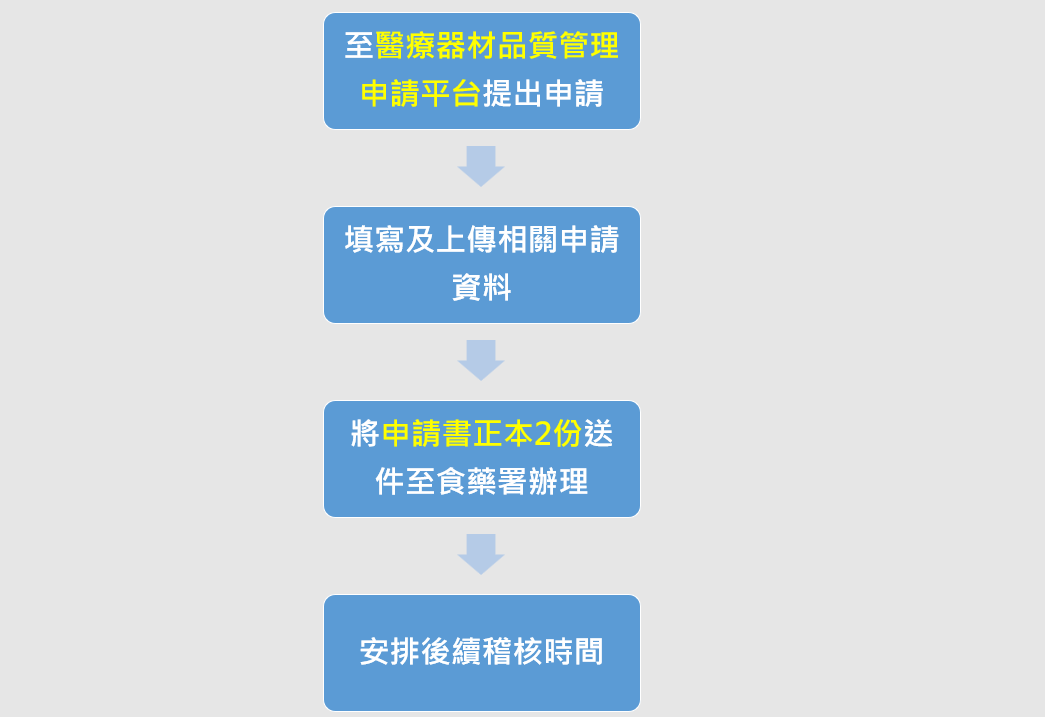
2.QMS申請方式:
使用醫療器材品質管理申請平臺https://mdqms.fda.gov.tw/Org/Login 提出申請,填寫及上傳相關申請資料,完成確認後下載申請書正本2份送件至食藥署辦理。詳細申請作業流程請參考國產醫療器材QMS稽核作業流程圖。
3.QMS後續查廠:
製造許可(認可登錄函)有效期為3年,若到期且繼續製造生產者,請申請後續查廠。按照醫療器材品質管理系統檢查及製造許可核發辦法第六條之規定,於證明文件有效期間屆滿之6個月前至12個月間主動提出申請,同樣填寫申請書並準備相關資料送件。後續檢查之許可範圍,得依業者之申請維持原許可範圍,亦可同時減少或增加項目,申請方式同樣填寫申請書並備齊相關資料送件。
4.QMS許可項目及作業內容變更:
依據醫療器材品質管理系統檢查及製造許可核發辦法第五條第一項第三款,許可項目及作業內容變更之申請及檢查程式準用該辦法第二條及第三條規定提出;申請方式同樣填寫申請書並備齊相關資料送件。
5.QMS申請費用:
依據醫療器材行政規費收費標準,如開具郵政匯票或即期支票,其抬頭為「衛生福利部食品藥物管理署」。
(1) 國內醫療器材品質管理系統檢查及其後續管理檢查:新臺幣六萬元。
(2) 醫療器材製造業者符合醫療器材品質管理系統準則第七十八條規定之檢查或其後續檢查:新臺幣三萬元。
6.QMS注意事項:
(1)TFDA於110年4月14日公告「醫療器材品質管理系統準則(QMS)」同時於110年5月1日與「醫療器材管理法」生效。醫療器材製造業者於110年4月30日前可申請GMP申請案(新案/後續案),此GMP申請案其核准之製造許可效期至113年4月30日止。但自110年5月1日起,新申請案應提出QMS申請,原有醫材製造業者(後續案)亦應符合並申請QMS,所有醫材製造業者最遲於113年4月30日前應符合QMS準則。
(2)依據醫療器材品質管理系統準則(QMS)第78條,製造業者僅生產附表所列醫療器材品項者,應就其生產之每一類型或系列分別建檔,並實施紀錄管制、申訴處理及矯正與預防措施。前項業者,除第十一條至第十三條(醫療器材檔案、檔管制、紀錄管制)、第四十七條(生產與服務提供之管制)、第五十五條(追溯性)、第六十三條(申訴處理)、第六十四條(通報、矯正預防措施、通知、回收)、第六十九條(產品之監管與量測)、第七十六條(矯正措施)及第七十七條(預防措施)規定外,不適用第二章至前章之規定。附表:https://law.moj.gov.tw/LawClass/LawGetFile.ashx?FileId=0000289844&lan=C
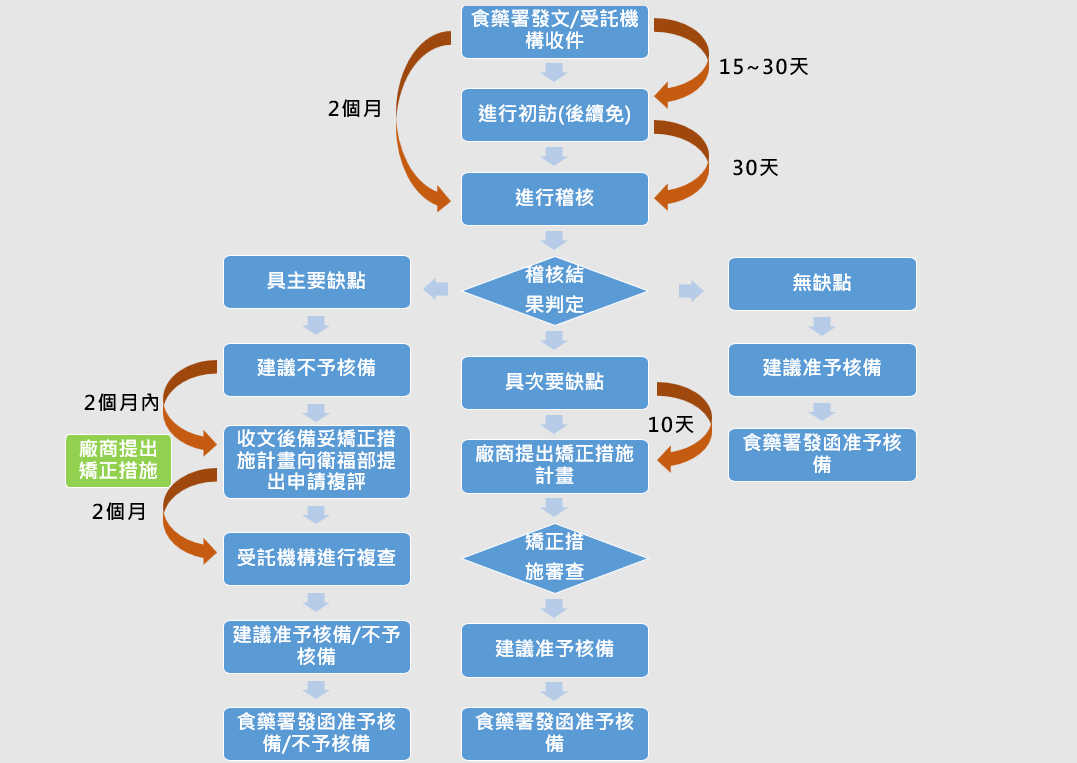
(3)國產製造業者於QMS稽核作業中,若發現主要缺點1項(含)以上或 10 項(含)以上次要缺點,該申請案將判定為不符合(複評申請參照檢查辦法第 3 條)。
Source : TFDA
ISO 13485 申請代辦顧問輔導
1.ISO 13485醫療器材品質管理系統之驗證為全球大部分對醫材上市審核的重要環節,其最新版 (2016) 增加風險管理、品質系統軟體確效、產品清潔、安裝、醫材產品相關服務、製程確效、滅菌確效、醫療器材軟體生命週期管理、植入式醫材追朔、與上市後監督與通報等醫材產業的特別條款。國際上歐盟 MDR(加嚴)、TFDA QMS(參照)、美國FDA QMSR(部分差異)、東南亞等地須國家都要求醫材製造業者必須符合近似ISO 13485:2016之品質管理系統。
現行歐盟醫療器材法規(Medical Devices Regulation, MDR)之CE認證過程,雖然取得ISO 13485驗證仍未完全符合歐盟醫療器材法規(MDR)取得醫材CE標誌的條件,因此,2021年已發布EN ISO 13485:2016+A11:2021,該標準修正包含了新的附錄ZA和ZB,將MDR和IVDR的要求與標準的具體條文結合。因此,符合ISO 13485視為符合歐盟MDR之CE標誌的基本醫材品質系統要求。
2.ISO 13485 :2016 證書申請之驗證機構(CB)與認證機構(AB)
ISO 13485之驗證機構(Certification Body,CB)是私人機構,包括 DNV、SGS、BSI、BV、TUV SUD、UL、DEKRA等。而國家認證機構(National Accreditation Body,AB)是政府機構,諸如中華民國TAF、美國 ANAB、英國UKAS、日本 JAB、、中國 CNAB、德國 DAR 及 韓國 KAB等機構,其位階等同驗證機構上級監督機構。
另,各國政府的認證機構(AB)共同成立國際認證論壇(International Accreditation Forum, IAF),陸續簽署多國相互承認協議,以減少驗證機構必須向各國政府的認證機構重覆申請認證。
3.ISO 13485之適用對象
醫療器材製造業者、創新研發公司、最終產品生產廠、關鍵零組件供應商、特殊製程提供者、滅菌服務提供者、售後服務提供者、倉儲物流服務提供者、經銷或販賣業者許可申請ISO 13485驗證。
4.符合ISO 13485 醫療器材品質系統管理標準之效益
(1) ISO 13485驗證可增加進入更多全球市場的渠道
(2)增加如何檢討並改善整個組織內的品質流程
(3)提升效率及降低成本,並監督整體供應鏈組織的績效
(4)證明貴司生產更安全有效的醫療器材
(5)符合法規要求與客戶要求
(6)提升製造醫材業者之品質聲譽
5.ISO 13485 :2016各主要章節
(1) 範圍 (Scope)
(2) 規範性引用 (Normative References)
(3) 名詞和定義 (Terms and definitions)
(4) 品質管理系統 (Quality management system)
(5) 管理責任 (Management responsibility)
(6) 資源管理 (Resource management)
(7) 產品實現 (Product realization)
(8) 量測、分析與改善 (Measurement, analysis and improvement)

領証顧問輔導優勢
領証之QMS與ISO 13485顧問具備QMS與ISO 13485之實務查廠豐沛經驗,亦具國際IRCA ISO 13485、ISO 14001、ISO 14064-1及ISO 14067主導稽核(查證)員之資格,具備多方領域之國際證照,可有效協助您熟悉醫材法規標準的要求,及建立完善環境及醫材品質管理系統(QMS及ISO 13485)。此外,我們能協助準備QMS及ISO 13485所需品質文件的輔導或撰寫,依循客戶特別須求而定,以客戶為導向是我們的職志。國外醫材輸入台灣販售一樣須符合醫療器材品質管理系統文件QSD ( Quality System Documentation),不同QSD申請模式對文件內容的要求亦有所差異,與原廠的溝通曠日廢時,導致許多醫材輸入業者在QSD申請過程中遇到很多的困難,我們專業醫材顧問以在TFDA的豐富審查經驗,協助您準備QSD所需的文件,亦可協助業者直接與原廠溝通QSD所需品質文件,以高效益行動力執行QSD申請代辦輔導服務,有效快速取得輸入醫材許可證。
TFDA QMS / QSD申請代辦顧問應檢附之文件:
1.依據醫療器材品質管理系統檢查及製造許可核發辦法(附表一)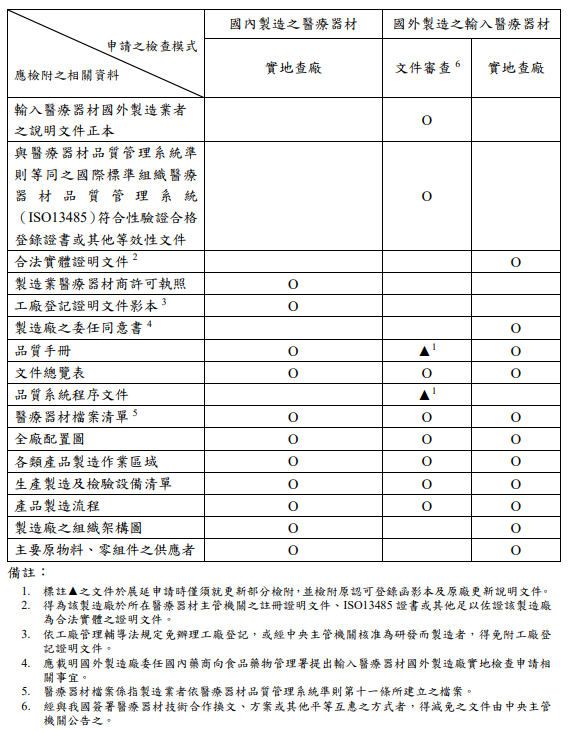
2.依據醫療器材品質管理系統檢查及製造許可核發辦法(附表二)
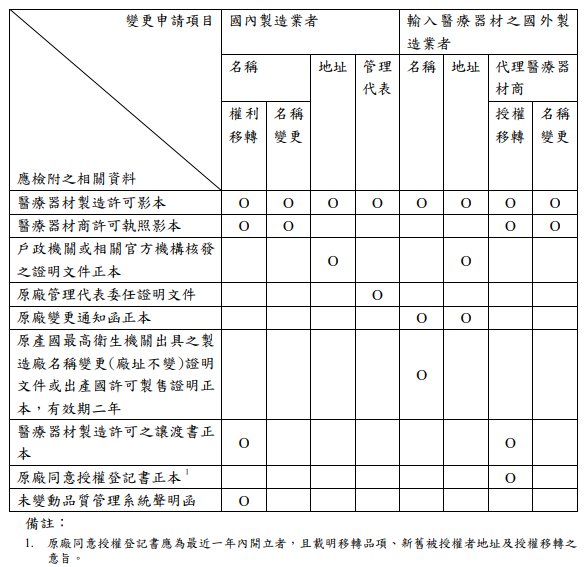
Source:TFDA

 加入領証生醫專頁
加入領証生醫專頁